Agile sequence analysis
Desktop programs for analyzing massively parallel sequence data
The Agile suite of programs allows the rapid analysis of clonal sequencing data, aligned to the human genome, with a view to identifying disease-causing sequence variants. It comprises several component programs, each with its own user guide and download page, as described below:
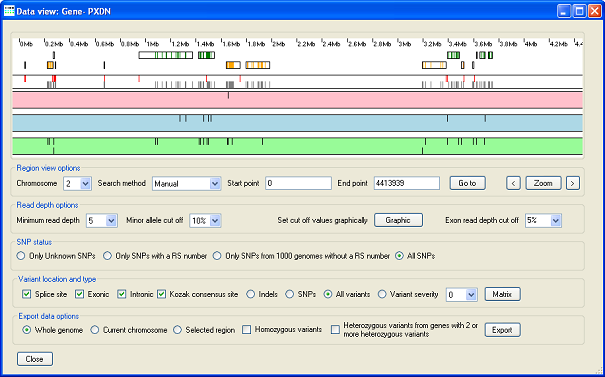
AgileVariantViewer identifying a PXDN variant that causes congenital cataract, corneal opacity, and glaucoma (citation).
Fastq and Fasta file manipulation and read quality filtering
AgilePairedEndReadsCombiner
Can combine paired end read sequences that overlap to form a single read and quality score string.
AgileQualityFilter
Allows the filtering out of low quality sequences, with the exported data formatted as either FASTA or FASTQ. If the reads are 5′‑end tagged with sample identifiers, the program will also sort and remove the tags from the output data.
Sam file manipulation
AgileSamFileSorter
Sorts the aligned sequence reads in an unordered SAM file by chromosome number and position.
AgileSamFileMerger
Combines two or more ordered SAM files to create a single ordered file.
Autozygosity mapping with exome data
AgileGenotyper
Creates a pseudo-microarray SNP genotyping file from an ordered SAM file containing exon sequence data. The file will contain the genotype data at over 0.5 million SNP sites previously identified by the 1000 Genomes project. Such a file can then be used as a data source for a mapping program designed for analyzing Affymetrix microarray SNP data (see Genetic mapping).
AgileMultiIdeogram
AgileMultiIdeogram displays autozygous regions from multiple individuals, identified in Affymetrix microarray SNP chip genotype data and/or exome variant data, against a circular ideogram of the human autosomal genome. AgileMultiIdeogram web page
AgileROH
AgileROH is the core functionality of AgileMultiIdeogram ported to C++ such that it can be run as a command line application in both Windows and Linux environments. The programs demonstrate how to use the source code to either just identify the autozygous regions and export to a text file or filter the VCF file to remove all variants not in the autozygous regions. AgileROH web page
AgileVariantMapper
Visualises sequence variant data from whole exome data, allowing the identification of autozygous regions in consanguineous individuals. The data can originate from files exported by AgileGenotyper, AgileAnnotator, AgileVariantViewer or a tab-delimited text file formatted as described in the user guide webpage.
AgileVCFMapper
AgileVCFMapper allows exome sequence variants in *.VCF files to be used to map disease loci in a similarly manner to AutoSNPa and IBDFinder. While it doesn't duplicate the functions of Phaser, Sample and DominantMapper, AgileVCFMapper can export the exome variant data to a coherent set of SNP genotype files that these programs can use.
Genome annotation file creation
AgileGAFCreator
AgileGAFCreator is designed to create annotation files of the human genome that can be used by other programs in Agile suite of Next Generation Sequencing programs.
Germline mutation detection
AgileAnnotator
Reads an ordered SAM file and identifies any sequence variant located in a protein-coding exon or within 50 bp of a splice site.
AgileKnownSNPFilter
Analyzes sequence variants exported by AgileAnnotator and identifies those previously found in the 1000 Genomes project.
AgileFileConverter
Reformats data in tab-delimited text files to a format that can be imported into AgileVariantViewer or AgileFileViewer. Since many commercial NGS service providers supply variant data in tab-delimited text files, AgileFileConverter allows such data to be analyzed by the Agile suite of programs.
AgileVariantViewer
Allows variants identified by AgileAnnotator and optionally filtered by AgileKnownSNPFilter to be interactively filtered by read depth and by allele read depth ratio. AgileVariantViewer can then export sequence variants for the whole genome, a single chromosome or a chromosomal region.
AgileGeneFilter
Allows sequence variants exported from AgileVariantViewer to be filtered, by identifying the genes within which they are located, and then performing a textual data search on those genes, using information from UniProt.
AgileFileViewer
Reads a sequence variant file created by AgileAnnotator (or one filtered by either AgileKnownSNPFilter or AgileVariantViewer) and displays associated information for each variant.
AgileVariantSelector
Reads multiple VCF files or 'annotated VCF' like files and identifies variants which occur at least n times in the data from affected individuals and discounts any variant that appears in a normal individuals data set.
GeneTIER
replaces the knowledge-based inference traditionally used in candidate disease gene prioritization, instead using experimental data from tissue-specific gene expression datasets. It is implemented as a hosted web application, and may be found here.
OVA
is an online variant filtering and prioritisation application. Ontology Variant Analysis Tool can filter your VCF files on a wide array of criteria. Remaining genes are prioritisated based on their functional and phenotypic profile similarity to a user supplied phenotype. It is implemented as a hosted web application, and may be found here.
Filtering the output from AlamutHT or Pindel
AgileExomeFilter
Enables the rapid filtering, sorting and screening of single-base variants as well as small indels derived from an exome-sequencing experiment and annotated by AlamutHT, allowing the rapid detection of possibly deleterious variants.
AgilePindelFilter
Enables the rapid filtering and screening of indel variants derived from an exome-sequencing experiment and annotated by Pindel, to allow the rapid detection of possibly deleterious variants.
Somatic mutation detection
AgileSMPoint
Identifies somatic sequence variants occurring at specific genomic positions and uses unaligned next-generation sequence data.
AgileSMAll
Identifies somatic sequence variants occurring at all positions within a PCR product and uses unaligned next-generation sequence data.